34 KiB
nf-core/taxprofiler: Output
Introduction
This document describes the output produced by the pipeline. Most of the plots are taken from the MultiQC report, which summarises results at the end of the pipeline.
The directories listed below will be created in the results directory after the pipeline has finished. All paths are relative to the top-level results directory.
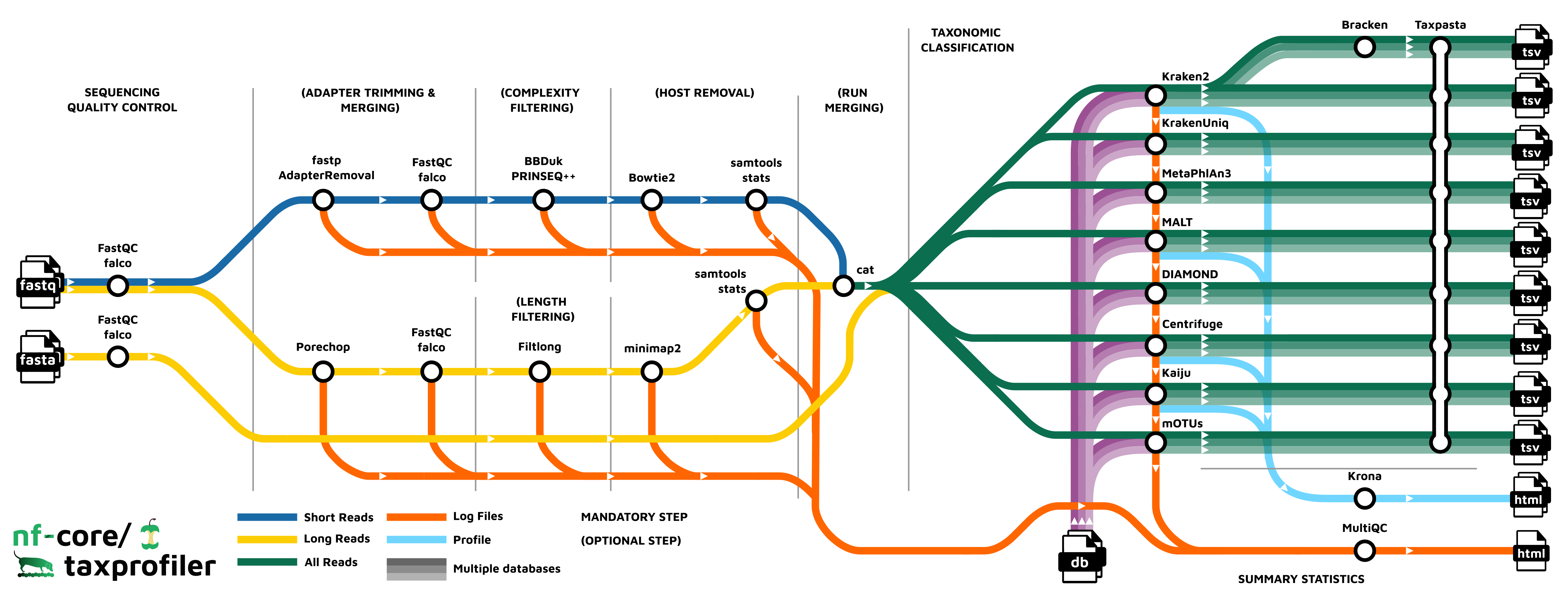
Pipeline overview
The pipeline is built using Nextflow and processes data using the following steps:
- FastQC - Raw read QC
- falco - Alternative to FastQC for raw read QC
- fastp - Adapter trimming for Illumina data
- AdapterRemoval - Adapter trimming for Illumina data
- Porechop - Adapter removal for Oxford Nanopore data
- BBDuk - Quality trimming and filtering for Illumina data
- PRINSEQ++ - Quality trimming and filtering for Illunina data
- Filtlong - Quality trimming and filtering for Nanopore data
- Bowtie2 - Host removal for Illumina reads
- minimap2 - Host removal for Nanopore reads
- SAMtools stats - Statistics from host removal
- SAMtools bam2fq - Converts unmapped BAM file to fastq format (minimap2 only)
- Bracken - Taxonomic classifier using k-mers and abundance estimations
- Kraken2 - Taxonomic classifier using exact k-mer matches
- KrakenUniq - Taxonomic classifier that combines the k-mer-based classification and the number of unique k-mers found in each species
- Centrifuge - Taxonomic classifier that uses a novel indexing scheme based on the Burrows-Wheeler transform (BWT) and the Ferragina-Manzini (FM) index.
- Kaiju - Taxonomic classifier that finds maximum (in-)exact matches on the protein-level.
- Diamond - Sequence aligner for protein and translated DNA searches.
- MALT - Sequence alignment and analysis tool designed for processing high-throughput sequencing data, especially in the context of metagenomics
- MetaPhlAn3 - Genome-level marker gene based taxonomic classifier
- mOTUs - Tool for marker gene-based OTU (mOTU) profiling.
- TAXPASTA - Tool to standardise taxonomic profiles as well as merge profiles across samples from the same database and classifier/profiler.
- MultiQC - Aggregate report describing results and QC from the whole pipeline
- Pipeline information - Report metrics generated during the workflow execution
FastQC or Falco
Output files
{fastqc,falco}/- {raw,preprocessed}
*html: FastQC or Falco report containing quality metrics in HTML format.*.txt: FastQC or Falco report containing quality metrics in TXT format.*.zip: Zip archive containing the FastQC report, tab-delimited data file and plot images (FastQC only).
- {raw,preprocessed}
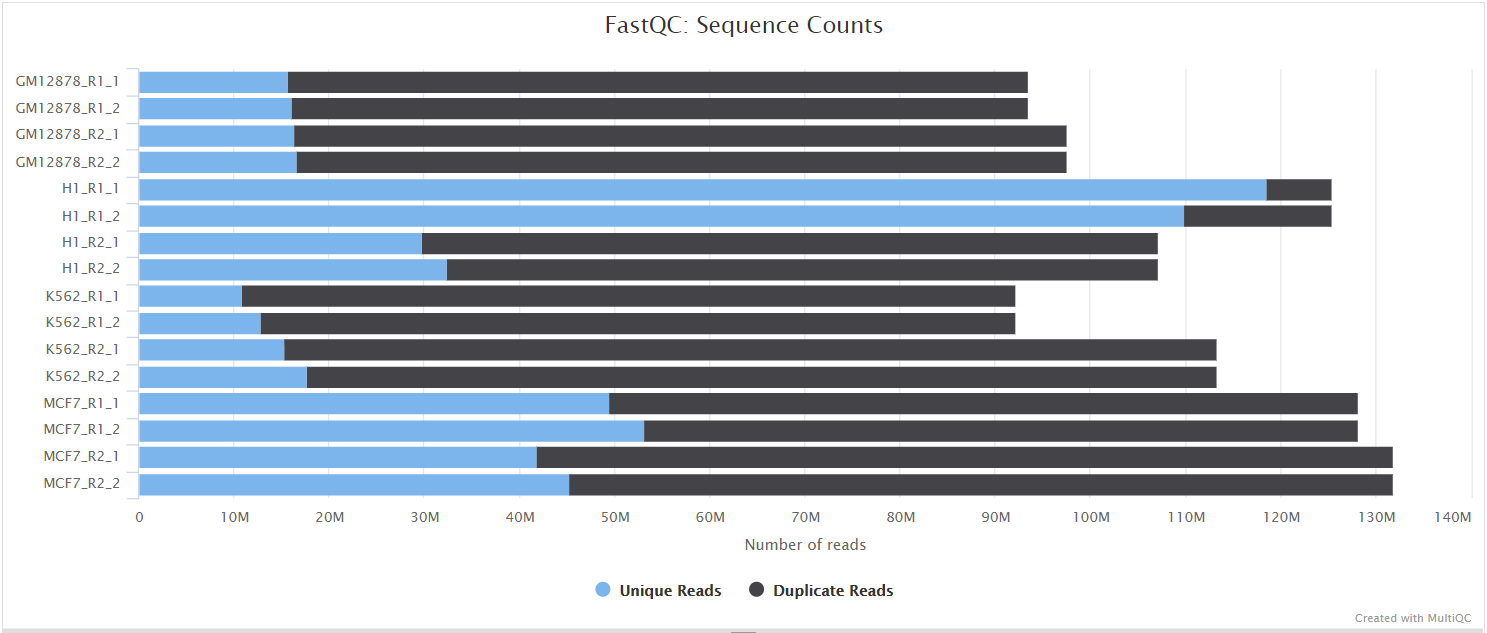
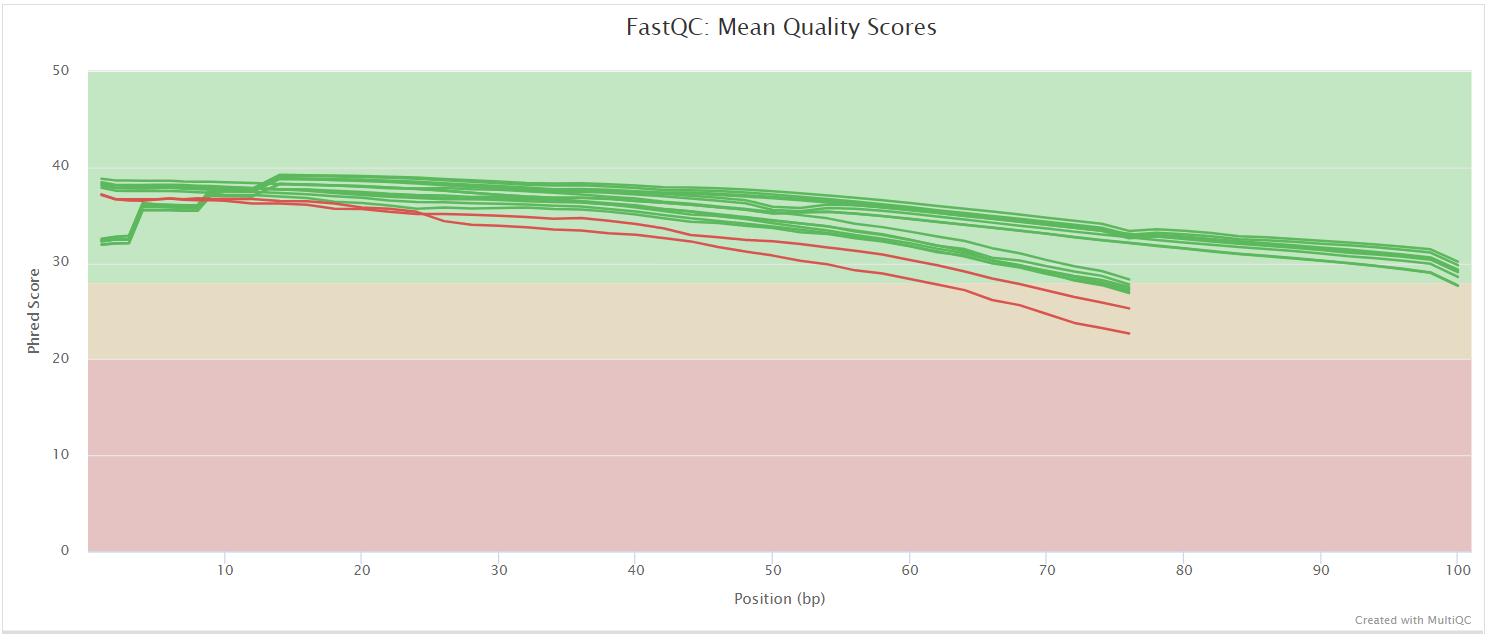
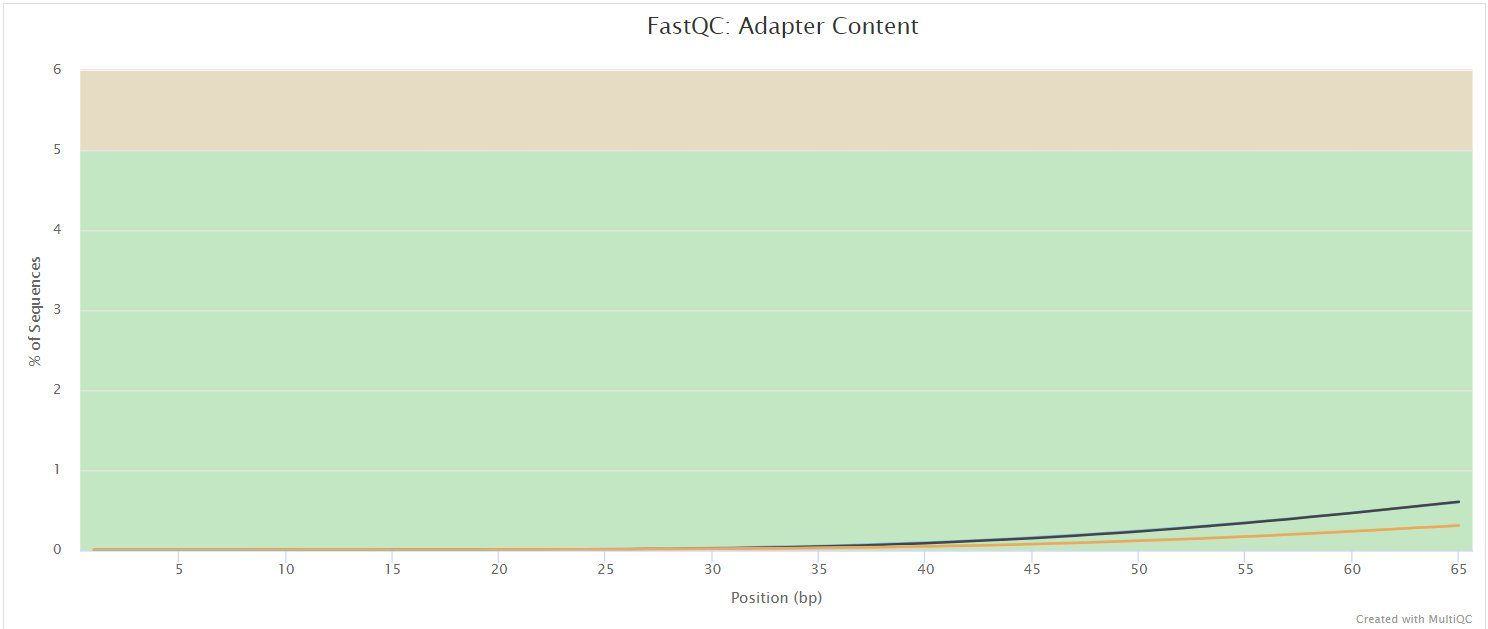
FastQC gives general quality metrics about your sequenced reads. It provides information about the quality score distribution across your reads, per base sequence content (%A/T/G/C), adapter contamination and overrepresented sequences. For further reading and documentation see the FastQC help pages.
If preprocessing is turned on, nf-core/taxprofiler runs FastQC/Falco twice -once before and once after adapter removal/read merging, to allow evaluation of the performance of these preprocessing steps. Note in the General Stats table, the columns of these two instances of FastQC/Falco are placed next to each other to make it easier to evaluate. However, the columns of the actual preprocessing steps (i.e, fastp, AdapterRemoval, and Porechop) will be displayed after the two FastQC/Falco columns, even if they were run 'between' the two FastQC/Falco jobs in the pipeline itself.
ℹ️ Falco produces identical output to FastQC but in the
falco/directory.
fastp
fastp is a FASTQ pre-processing tool for quality control, trimmming of adapters, quality filtering and other features.
It is used in nf-core/taxprofiler for adapter trimming of short-reads.
Output files
fastp<sample_id>.fastp.fastq.gz: File with the trimmed unmerged fastq reads.<sample_id>.merged.fastq.gz: File with the reads that were successfully merged.<sample_id>.*{log,html,json}: Log files in different formats.
By default nf-core/taxprofiler will only provide the <sample_id>.fastp.fastq.gz file if fastp is selected. The file <sample_id>.merged.fastq.gz will be available in the output folder if you provide the argument --shortread_qc_mergepairs (optionally retaining un-merged pairs when in combination with --shortread_qc_includeunmerged).
You can change the default value for low complexity filtering by using the argument --shortread_complexityfilter_fastp_threshold.
AdapterRemoval
AdapterRemoval searches for and removes remnant adapter sequences from High-Throughput Sequencing (HTS) data and (optionally) trims low quality bases from the 3' end of reads following adapter removal. It is popular in the field of palaeogenomics. The output logs are stored in the results folder, and as a part of the MultiQC report.
Output files
adapterremoval/<sample_id>.settings: AdapterRemoval log file containing general adapter removal, read trimming and merging statistics<sample_id>.collapsed.fastq.gz- read-pairs that merged and did not undergo trimming (only when--shortread_qc_mergepairssupplied)<sample_id>.collapsed.truncated.fastq.gz- read-pairs that merged underwent quality trimming (only when--shortread_qc_mergepairssupplied)<sample_id>.pair1.truncated.fastq.gz- read 1 of pairs that underwent quality trimming<sample_id>.pair2.truncated.fastq.gz- read 2 of pairs that underwent quality trimming (and could not merge if--shortread_qc_mergepairssupplied)<sample_id>.singleton.truncated.fastq.gz- orphaned read pairs where one of the pair was discarded<sample_id>.discard.fastq.gz- reads that were discarded due to length or quality filtering
By default nf-core/taxprofiler will only provide the .settings file if AdapterRemoval is selected.
You will only find the .fastq files in the results directory if you provide --save_preprocessed_reads. If this is selected, you may receive different combinations of .fastq files for each sample depending on the input types - e.g. whether you have merged or not, or if you're supplying both single- and paired-end reads.
⚠️ The resulting
.fastqfiles may not always be the 'final' reads that go into taxprofiling, if you also run other steps such as complexity filtering, host removal, run merging etc..
Porechop
Porechop is a tool for finding and removing adapters from Oxford Nanopore reads. Adapters on the ends of reads are trimmed and if a read has an adapter in its middle, it is considered a chimeric and it chopped into separate reads.
Output files
porechop<sample_id>.log: Log file containing trimming statistics<sample_id>.fastq.gz: Adapter-trimmed file
The output logs are saved in the output folder and are part of MultiQC report.You do not normally need to check these manually.
You will only find the .fastq files in the results directory if you provide --save_preprocessed_reads.
⚠️ We do not recommend using Porechop if you are already trimming the adapters with ONT's basecaller Guppy.
BBDuk
BBDuk stands for Decontamination Using Kmers. BBDuk was developed to combine most common data-quality-related trimming, filtering, and masking operations into a single high-performance tool.
It is used in nf-core/taxprofiler for complexity filtering using different algorithms. This means that it will remove reads with low sequence diversity (e.g. mono- or dinucleotide repeats).
Output files
bbduk/<sample_id>.bbduk.log: log file containing filtering statistics<sample_id>.fastq.gz: resulting FASTQ file without low-complexity reads
By default nf-core/taxprofiler will only provide the .log file if BBDuk is selected as the complexity filtering tool. You will only find the complexity filtered reads in your results directory if you provide --save_complexityfiltered_reads .
⚠️ The resulting
.fastqfiles may not always be the 'final' reads that go into taxprofiling, if you also run other steps such as host removal, run merging etc..
PRINSEQ++
PRINSEQ++ is a C++ implementation of the prinseq-lite.pl program. It can be used to filter, reformat or trim genomic and metagenomic sequence data.
It is used in nf-core/taxprofiler for complexity filtering using different algorithms. This means that it will remove reads with low sequence diversity (e.g. mono- or dinucleotide repeats).
Output files
prinseqplusplus/<sample_id>.log: log file containing number of reads. Row IDs correspond to:min_len, max_len, min_gc, max_gc, min_qual_score, min_qual_mean, ns_max_n, noiupac, derep, lc_entropy, lc_dust, trim_tail_left, trim_tail_right, trim_qual_left, trim_qual_right, trim_left, trim_right<sample_id>_good_out.fastq.gz: resulting FASTQ file without low-complexity reads
By default nf-core/taxprofiler will only provide the .log file if PRINSEQ++ is selected as the complexity filtering tool. You will only find the complexity filtered .fastq files in your results directory if you supply --save_complexityfiltered_reads .
⚠️ The resulting
.fastqfiles may not always be the 'final' reads that go into taxprofiling, if you also run other steps such as host removal, run merging etc..
Filtlong
Filtlong is a quality filtering tool for long reads. It can take a set of small reads and produce a smaller, better subset.
Output files
filtlong<sample_id>_filtered.fastq.gz: Quality or short read data filtered file<sample_id>_filtered.log: log file containing summary statistics
You will only find the .fastq files in the results directory if you provide --save_preprocessed_reads.
⚠️ We do not recommend using Filtlong if you are performing filtering of low quality reads with ONT's basecaller Guppy.
Bowtie2
Bowtie 2 is an ultrafast and memory-efficient tool for aligning sequencing reads to long reference sequences. It is particularly good at aligning reads of about 50 up to 100s or 1,000s of characters, and particularly good at aligning to relatively long (e.g. mammalian) genomes.
It is used with nf-core/taxprofiler to allow removal of 'host' (e.g. human) and/or other possible contaminant reads (e.g. Phi X) from short-read .fastq files prior to profiling.
Output files
bowtie2/build/*.bt2: Bowtie2 indicies of reference genome, only if--save_hostremoval_indexsupplied.
align/<sample_id>.bam: BAM file containing reads that aligned against the user-supplied reference genome as well as unmapped reads<sample_id>.bowtie2.log: log file about the mapped reads<sample_id>.unmapped.fastq.gz: the off-target reads from the mapping that is used in downstream steps.
By default nf-core/taxprofiler will only provide the .log file if host removal is turned on. You will only have a .bam file if you specify --save_hostremoval_bam. This will contain both mapped and unmapped reads. You will only get FASTQ files if you specify to save --save_hostremoval_unmapped - these contain only unmapped reads.
ℹ️ Unmapped reads in FASTQ are only found in this directory for short-reads, for long-reads see
samtools/bam2fq/
⚠️ The resulting
.fastqfiles may not always be the 'final' reads that go into taxprofiling, if you also run other steps such as run merging etc..
ℹ️ While there is a dedicated section in the MultiQC HTML for Bowtie2, these values are not displayed by default in the General Stats table. Rather, alignment statistics to host genome is reported via samtools stats module in MultiQC report for direct comparison with minimap2 (see below).
minimap2
minimap2 is an alignment tool suited to mapping long reads to reference sequences.
It is used with nf-core/taxprofiler to allow removal of 'host' (e.g. human) or other possible contaminant reads from long-read .fastq files prior to taxonomic classification/profiling.
Output files
minimap2build/*.mmi2: minimap2 indices of reference genome, only if--save_hostremoval_indexsupplied.
align/<sample_id>.bam: Alignment file in BAM format containing both mapped and unmapped reads.
By default, nf-core/taxprofiler will only provide the .bam file containing mapped and unmapped reads if saving of host removal for long reads is turned on via --save_hostremoval_bam.
ℹ️ minimap2 is not yet supported as a module in MultiQC and therefore there is no dedicated section in the MultiQC HTML. Rather, alignment statistics to host genome is reported via samtools stats module in MultiQC report.
ℹ️ Unlike Bowtie2, minimap2 does not produce an unmapped FASTQ file by itself. See
samtools/bam2fq
SAMtools bam2fq
SAMtools bam2fq converts a .sam, .bam, or .cram alignment file to FASTQ format
Output files
samtoolsstats<sample_id>_interleaved.fq.gz: Unmapped reads only in FASTQ gzip format
This directory will be present and contain the unmapped reads from the .fastq format from long-read minimap2 host removal, if --save_hostremoval_unmapped is supplied
ℹ️ For short-read unmapped reads, see bowtie2.
SAMtools stats
SAMtools stats collects statistics from a .sam, .bam, or .cram alignment file and outputs in a text format.
Output files
samtools/stats<sample_id>.stats: File containing samtools stats output.
In most cases you do not need to check this file, as it is rendered in the MultiQC run report.
Run Merging
nf-core/taxprofiler offers the option to merge FASTQ files of multiple sequencing runs or libraries that derive from the same sample, as specified in the input samplesheet.
This is the last preprocessing step, so if you have multiple runs or libraries (and run merging turned on), this will represent the final reads that will go into classification/profiling steps.
Output files
run_merging/*.fastq.gz: Concatenated FASTQ files on a per-sample basis
Note that you will only find samples that went through the run merging step in this directory. For samples that had a single run or library will not go through this step of the pipeline and thus will not be present in this directory.
⚠️ You must make sure to turn on the saving of the reads from the previous preprocessing step you may have turned on, if you have single-run or library reads in your pipeline run, and wish to save the final reads that go into classification/profiling!
Bracken
Bracken (Bayesian Reestimation of Abundance with Kraken) is a highly accurate statistical method that computes the abundance of species in DNA sequences from a metagenomics sample. Braken uses the taxonomy labels assigned by Kraken, a highly accurate metagenomics classification algorithm, to estimate the number of reads originating from each species present in a sample.
🛈 The first step of using Bracken requires running Kraken2, therefore the initial results before abundance estimation will be found in
<your_results>/kraken2/<your_bracken_db_name>.
Output files
bracken/bracken_<db_name>_combined_reports.txt: combined bracken results as output from Bracken'scombine_bracken_outputs.pyscript<db_name>/<sample>_<db_name>.tsv: TSV file containing per-sample summary of Bracken results with abundance information
The main taxonomic profiling file from Bracken is the *.tsv file. This provides the basic results from Kraken2 but with the corrected abundance information.
Kraken2
Kraken is a taxonomic sequence classifier that assigns taxonomic labels to DNA sequences. Kraken examines the k-mers within a query sequence and uses the information within those k-mers to query a database. That database maps -mers to the lowest common ancestor (LCA) of all genomes known to contain a given k-mer.
Output files
kraken2/<db_name>_combined_reports.txt: A combined profile of all samples aligned to a given database (as generated bykrakentools)<db_name>/<sample_id>_<db_name>.classified.fastq.gz: FASTQ file containing all reads that had a hit against a reference in the database for a given sample<sample_id>_<db_name>.unclassified.fastq.gz: FASTQ file containing all reads that did not have a hit in the database for a given sample<sample_id>_<db_name>.report.txt: A Kraken2 report that summarises the fraction abundance, taxonomic ID, number of Kmers, taxonomic path of all the hits in the Kraken2 run for a given sample<sample_id>_<db_name>.classifiedreads.txt: A list of read IDs and the hits each read had against each database for a given sample
The main taxonomic classification file from Kraken2 is the _combined_reports.txt or *report.txt file. The former provides you the broadest over view of the taxonomic classification results across all samples against a single databse, where you get two columns for each sample e.g. 2_all and 2_lvl, as well as a summarised column summing up across all samples tot_all and tot_lvl. The latter gives you the most information for a single sample. The report file is also used for the taxpasta step.
You will only receive the .fastq and *classifiedreads.txt file if you supply --kraken2_save_reads and/or --kraken2_save_readclassification parameters to the pipeline.
KrakenUniq
KrakenUniq (formerly KrakenHLL) is an extenson to the fast k-mer-based classification Kraken with an efficient algorithm for additionally assessing the coverage of unique k-mers found in each species in a dataset.
Output files
krakenuniq/<db_name>/<sample_id>_<db_name>.classified.fastq.gz: FASTQ file containing all reads that had a hit against a reference in the database for a given sample<sample_id>_<db_name>.unclassified.fastq.gz: FASTQ file containing all reads that did not have a hit in the database for a given sample<sample_id>_<db_name>.report.txt: A Kraken2-style report that summarises the fraction abundance, taxonomic ID, number of Kmers, taxonomic path of all the hits, with an additional column for k-mer coverage, that allows for more accurate distinguishing between false-positive/true-postitive hits<sample_id>_<db_name>.classifiedreads.txt: A list of read IDs and the hits each read had against each database for a given sample
The main taxonomic classification file from KrakenUniq is the *report.txt file. This is an extension of the Kraken2 report with the additional k-mer coverage information that provides more information about the accuracy of hits.
You will only receive the .fastq and *classifiedreads.txt file if you supply --krakenuniq_save_reads and/or --krakenuniq_save_readclassification parameters to the pipeline.
⚠️ The output system of KrakenUniq can result in other
stdoutorstderrlogging information being saved in the report file, therefore you must check your report files before downstream use!
Centrifuge
Centrifuge is a taxonomic sequence classifier that uses a Burrows-Wheeler transform and Ferragina-Manzina index for storing and mapping sequences.
Output files
centrifuge<sample_id>.centrifuge.mapped.fastq.gz:FASTQfiles containing all mapped reads<sample_id>.centrifuge.report.txt: A classification report that summarises the taxonomic ID, the taxonomic rank, length of genome sequence, number of classified and uniquely classified reads<sample_id>.centrifuge.results.txt: A file that summarises the classification assignment for a read, i.e read ID, sequence ID, score for the classification, score for the next best classification, number of classifications for this read<sample_id>.centrifuge.txt: A Kraken2-style report that summarises the fraction abundance, taxonomic ID, number of k-mers, taxonomic path of all the hits in the centrifuge run for a given sample<sample_id>.centrifuge.unmapped.fastq.gz: FASTQ file containing all unmapped reads
The main taxonomic classification files from Centrifuge are the _combined_reports.txt, *report.txt, *results.txt and the *centrifuge.txt. The latter is used by the taxpasta step. You will receive the .fastq files if you supply --centrifuge_save_reads.
Kaiju
Kaiju is a taxonomic classifier that finds maximum exact matches on the protein-level using the Burrows-Wheeler transform.
Output files
kaijukaiju_<db_name>_combined_reports.txt: A combined profile of all samples aligned to a given database (as generated by kaiju2table)<db_name>/<sample_id>_<db_name>.kaiju.tsv: Raw output from Kaiju with taxonomic rank, read ID and taxonic ID<sample_id>_<db_name>.kaijutable.txt: Summarised Kaiju output with fraction abundance, taxonomic ID, number of reads, and taxonomic names (as generated bykaiju2table)
The most useful summary file is the _combined_reports.txt file which summarises hits across all reads and samples. Separate per-sample versions summaries can be seen in <db>/*.txt. However if you wish to look at more precise information on a per-read basis, see the *tsv file. The default taxonomic rank is species. You can provide a different one by updating the argument --kaiju_taxon_rank.
DIAMOND
DIAMOND is a sequence aligner for translated DNA searches or protein sequences against a protein reference database such as NR. It is a replacement for the NCBI BLAST software tools.It has many key features and it is used as taxonomic classifier in nf-core/taxprofiler.
Output files
diamond<sample_id>.log: A log file containing stdout information<sample_id>*.{blast,xml,txt,daa,sam,tsv,paf}: A file containing alignment information in various formats, or taxonomic information in a text-based format. Exact output depends on user choice.
By default you will receive a TSV output. Alternatively, you will receive a *.sam file if you provide the parameter --diamond_save_reads but in this case no taxonomic classification will be available(!), only the aligned reads in sam format.
ℹ️ DIAMOND has many output formats, so depending on your choice with
--diamond_output_formatyou will receive the taxonomic information in a different format.
MALT
MALT is a fast replacement for BLASTX, BLASTP and BLASTN, and provides both local and semi-global alignment capabilities.
Output files
malt/<db_name>/<sample_id>.blastn.sam: sparse SAM file containing alignments of each hit<sample_id>.megan: summary file that can be loaded into the MEGAN6 interactive viewer. Generated by MEGAN6 companion toolrma2info<sample_id>.rma6: binary file containing all alignments and taxonomic information of hits that can be loaded into the MEGAN6 interactive viewer<sample_id>.txt.gz: text file containing taxonomic IDs and read counts against each taxon. Generated by MEGAN6 companion toolrma2info
The main output of MALT is the .rma6 file format, which can be only loaded into MEGAN and it's related tools. We provide the rma2info text files for improved compatibility with spreadsheet programs and other programmtic data manipulation tools, however this has only limited information compared to the 'binary' RMA6 file format (the .txt file only contains taxonomic ID and count, whereas RMA6 has taxonomic lineage information).
You will only receive the .sam and .megan files if you supply --malt_save_reads and/or --malt_generate_megansummary parameters to the pipeline.
MetaPhlAn3
MetaPhlAn3 is a computational tool for profiling the composition of microbial communities (Bacteria, Archaea and Eukaryotes) from metagenomic shotgun sequencing data (i.e. not 16S) with species-level resolution via marker genes.
Output files
metaphlan3/metaphlan3_<db_name>_combined_reports.txt: A combined profile of all samples aligned to a given database (as generated bymetaphlan_merge_tables)<db_name>/<sample_id>.biom: taxonomic profile in BIOM format<sample_id>.bowtie2out.txt: BowTie2 alignment information (can be re-used for skipping alignment when re-running MetaPhlAn3 with different parameters)<sample_id>_profile.txt: MetaPhlAn3 taxonomic profile including abundance estimates
The main taxonomic profiling file from MetaPhlAn3 is the *_profile.txt file. This provides the abundance estimates from MetaPhlAn3 however does not include raw counts by default.
mOTUs
mOTUS is a taxonomic profiler that maps reads to a unique marker specific database and estimates the relative abundance of known and unknown species.
Output files
motus<sample_id>.log: A log file that contains summary statistics<sample_id>.out: A classification file that summarises taxonomic identifiers, by default at the rank of mOTUs (i.e., species level), and their relative abundances in the profiled sample.motus_<db_name>_combined_reports.txt: A combined profile of all samples aligned to a given database (as generated bymotus_merge)
Normally *_combined_reports.txt is the most useful file for downstream analyses, but the per sample .out file can provide additional more specific information. By default, nf-core/taxprofiler is providing a column describing NCBI taxonomic ID as this is used in the taxpasta step. You can disable this column by activating the argument --motus_remove_ncbi_ids.
You will receive the relative abundance instead of read counts if you provide the argument --motus_use_relative_abundance.
Krona
Krona allows the exploration of (metagenomic) hierarchical data with interactive zooming, multi-layered pie charts.
Krona charts will be generated by the pipeline for supported tools (Kraken2, Centrifuge, Kaiju, and MALT)
Output files
krona/<tool_name>_<db_name>.html: per-tool/per-database interactive HTML file containing hierarchical piecharts
The resulting HTML files can be loaded into your web browser for exploration. Each file will have a dropdown to allow you to switch between each sample aligned against the given database of the tool.
TAXPASTA
TAXPASTA standardises and merges two or more taxonomic profiles across samples into one single table. It supports multiple different classifiers simplifying comparison of taxonomic classification results between tools and databases.
Output files
-
taxpasta<tool>_<database>*.{tsv,csv,arrow,parquet,biom}: Standardised taxon table containing multiple samples. The standard format is thetsv. The first column describes the taxonomy ID and the rest of the columns describe the read counts for each sample.
By providing the path to a directory containing taxdump files to --taxpasta_taxonomy_dir, the taxon name, the taxon rank, the taxon's entire lineage including taxon names and/or the taxon's entire lineage including taxon identifiers can also be added in the output in addition to just the taxon ID. Addition of this extra information can be turned by using the parameters --taxpasta_add_name, --taxpasta_add_rank, --taxpasta_add_lineage and --taxpasta_add_idlineage respectively.
These files will likely be the most useful files for the comparison of differences in classification between different tools or building consensuses, with the caveat they have slightly less information than the actual output from each tool (which may have non-standard information e.g. taxonomic rank, percentage of hits, abundance estimations).
The following report files are used for the taxpasta step:
- Bracken:
<sample>_<db_name>.tsvTaxpasta used thenew_est_readscolumn for the standardised profile. - Centrifuge:
<sample_id>.centrifuge.txtTaxpasta uses thedirect_assigned_readscolumn for the standardised profile. - Diamond:
<sample_id>Taxpasta summarises number of reads per NCBI taxonomy ID standardised profile. - Kaiju:
<sample_id>_<db_name>.kaijutable.txtTaxpasta uses thereadscolumn from kaiju2table standardised profile. - KrakenUniq:
<sample_id>_<db_name>.report.txtTaxpasta uses thereadscolumn for the standardised profile. - Kraken2:
<sample_id>_<db_name>.report.txtTaxpasta uses thedirect_assigned_readscolumn for the standardised profile. - MALT:
<sample_id>.txt.gzTaxpasta uses thecount(second) column from the output of MEGAN6's rma2info for the standardised profile. - MetaPhlAn3:
<sample_id>_profile.txtTaxpasta uses therelative_abundancecolumn multiplied with a fixed number to yield an integer for the standardised profile. - mOTUs:
<sample_id>.outTaxpasta uses theread_countcolumn for the standardised profile.
⚠️ Please aware the outputs of each tool's standardised profile may not be directly comparable between each tool. Some may report raw read counts, whereas others may report abundance information. Please always refer to the list above, for which information is used for each tool.
MultiQC
Output files
multiqc/multiqc_report.html: a standalone HTML file that can be viewed in your web browser.multiqc_data/: directory containing parsed statistics from the different tools used in the pipeline.multiqc_plots/: directory containing static images from the report in various formats.
MultiQC is a visualization tool that generates a single HTML report summarising all samples in your project. Most of the pipeline QC results are visualised in the report and further statistics are available in the report data directory.
Results generated by MultiQC collate pipeline QC from supported tools e.g. FastQC. The pipeline has special steps which also allow the software versions to be reported in the MultiQC output for future traceability. For more information about how to use MultiQC reports, see http://multiqc.info.
All tools in taxprofiler supported by MultiQC will have a dedicated section showing summary statistics of each tool based on information stored in log files.
You can expect in the MultiQC reports either sections and/or general stats columns for the following tools:
- fastqc
- adapterRemoval
- fastp
- bbduk
- prinseqplusplus
- porechop
- filtlong
- bowtie2
- minimap2
- samtools (stats)
- kraken
- bracken
- centrifuge
- kaiju
- metaphlan
- diamond
- malt
- motus
ℹ️ The 'General Stats' table by default will only show statistics referring to pre-processing steps, and will not display possible values from each classifier/profiler, unless turned on by the user within the 'Configure Columns' menu or via a custom MultiQC config file (
--multiqc_config)
Pipeline information
Output files
pipeline_info/- Reports generated by Nextflow:
execution_report.html,execution_timeline.html,execution_trace.txtandpipeline_dag.dot/pipeline_dag.svg. - Reports generated by the pipeline:
pipeline_report.html,pipeline_report.txtandsoftware_versions.yml. Thepipeline_report*files will only be present if the--email/--email_on_failparameter's are used when running the pipeline. - Reformatted samplesheet files used as input to the pipeline:
samplesheet.valid.csv.
- Reports generated by Nextflow:
Nextflow provides excellent functionality for generating various reports relevant to the running and execution of the pipeline. This will allow you to troubleshoot errors with the running of the pipeline, and also provide you with other information such as launch commands, run times and resource usage.